Individualized mRNA vaccines evoke durable T cell immunity in adjuvant TNBC
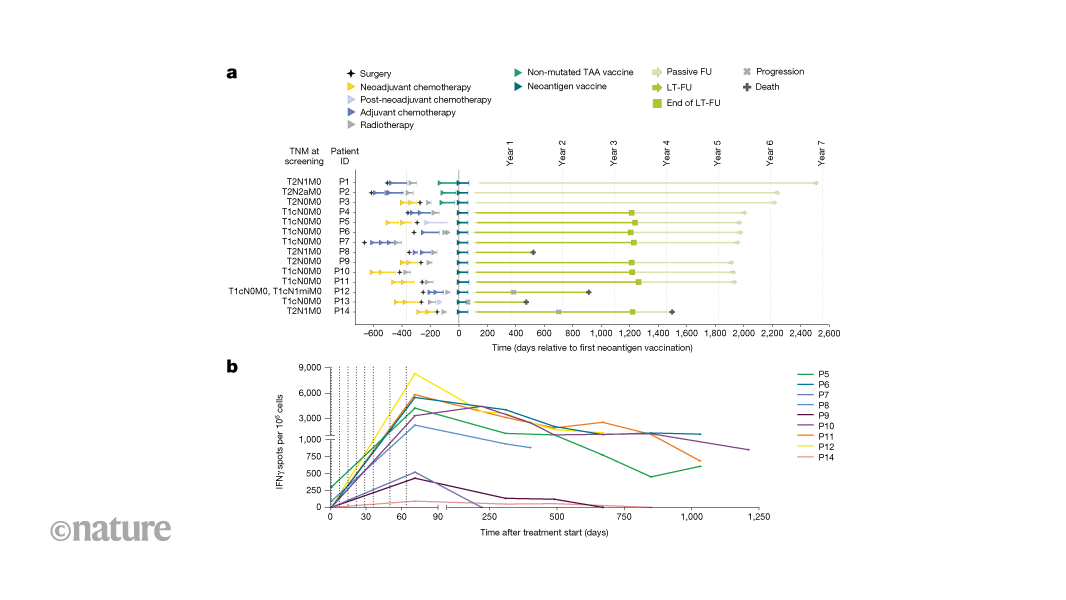
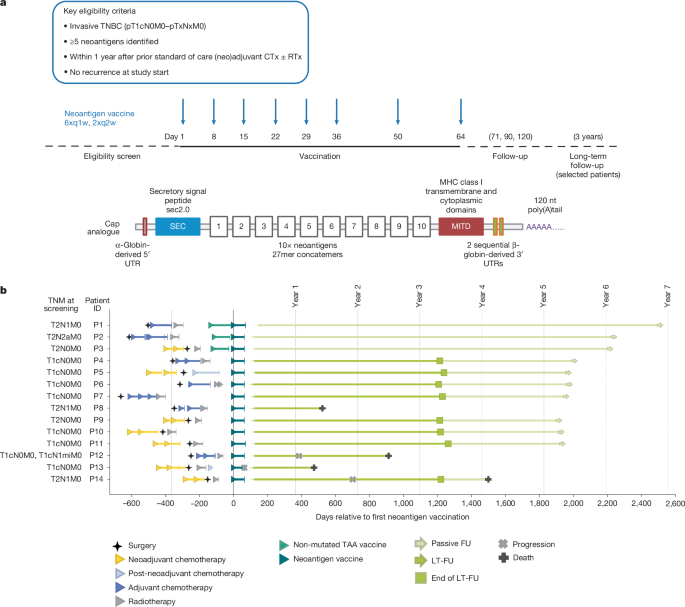
Trial design and data reportingThe primary objective of this open-label, first-in-human, phase 1, three-arm umbrella trial (ClinicalTrials.gov: NCT02316457) was to separately assess the feasibility, safety, and tolerability profile of two different mRNA–LPX-based vaccine types: an off-the-shelf warehouse vaccine composed of non-mutated TAAs and an on-demand manufactured individualized neoantigen vaccine. Vaccine-induced antigen-specific immune responses were investigated (secondary endpoint). Here, we report findings related to the individualized neoantigen vaccine arm of this trial. The trial was carried out in Germany and Sweden in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines, and with approval by the independent ethics committees (Ethik-Kommission of the Landesärztekammer Rheinland Pfalz, Mainz, Germany and Regionala Etikprövningsnämnden, Uppsala, Sweden) and the competent regulatory authority (Paul-Ehrlich Institute, Langen, Germany and Medical Products Agency, Uppsala, Sweden). All patients provided written informed consent.Eligibility criteria were: histologically confirmed invasive adenocarcinoma TNBC (pT1cN0M0–[any]T[any]NM0); previous standard of care treatment (that is, neoadjuvant chemotherapy of the primary tumour followed by surgery or surgery and adjuvant chemotherapy); prior radiotherapy was allowed; at least 18 years of age; adequate haematopoietic, hepatic and renal function; tumours expressing at least 5 neoantigens. Patients were eligible for enrolment within one year after completion of standard of care therapy per local policy (for example, surgery and/or chemotherapy and/or radiotherapy). Key exclusion criteria were the recurrence of breast cancer prior to the start of trial treatment and presence of clinically relevant autoimmune disease or active viral infections. The individualized neoantigen vaccine consisted of two single-stranded RNA molecules each encoding up to ten neoantigen vaccine targets selected based on somatic mutation analysis of each patient’s tumour. RNAs were liposomally formulated into RNA–LPX for intravenous administration as described11. For patients treated with neoadjuvant chemotherapy, the FFPE tumour sample from the diagnostic core biopsy was used for analysis of tumour antigen expression. In exceptional cases (for example, low sample quality), the resected FFPE tumour tissue from surgery could be used for analysis of antigen expression instead. agCapture 3.4.2.6 (ArisGlobal) was used for electronic data capture.The clinical trial report of the primary and secondary endpoints assessed in the main study phase until end of treatment and a 56-day follow-up period in 2020 was submitted to health authorities in spring 2021. Beyond this follow-up, the three patients pretreated with bridging TAA vaccine were followed up passively; one patient consented to provide blood samples via a research project. The following 11 patients (only treated with neoantigen vaccine) participated in an active long-term follow-up for 3 years until 2023. The data generated in the long-term follow-up have been summarized in an addendum to the clinical trial report and submitted to health authorities in the spring of 2024. Following this follow-up period, these patients were then followed up passively.Next generation sequencingTumour DNA was extracted from three 10 μm curls of FFPE tumour tissue in duplicates using a modified version of QIAamp DNA FFPE Tissue kit (Qiagen). RNA extraction was done in duplicates using the ExpressArt Clear FFPE RNAready from AmpTec. For DNA extractions from PBMC cells, the DNeasy Blood and Tissue Kit (Qiagen) was used. Extracted nucleic acids were used for generation of various NGS libraries. Targeted RNA-seq libraries were constructed in duplicate from FFPE tumour tissue RNA using the NEBNext RNA First Strand Synthesis Module and NEBNext Ultra Directional RNA Second Strand Synthesis Module for cDNA syntheses and a modified version of SureSelect XT V6 Human All Exon (Agilent) using 100 ng total RNA input. DNA whole-exome libraries were constructed in duplicates from 100 ng of FFPE tumour DNA and matching PBMC DNA using a modified version of SureSelect XT V6 Human All Exon (Agilent). NGS libraries for whole-exome sequencing of the tumour and matching PBMCs were prepared by fragmenting 100 ng genomic DNA in a total volume of 15 μl using microTUBE-15 AFA Beads Screw-Cap (Covaris) to an average fragment length of approximately 150 bp. For NGS, the libraries were diluted to 3 nM and clustered at 10 pM using the Illumina HiSeq 3000/4000 PE Cluster Kit. Pooled exome library from FFPE and PBMC DNA were sequenced as 4-plexes on three lanes, whereas the RNA library replicates were sequenced as 2-plexes in one lane. All libraries were sequenced paired-end 50 nt on an Illumina HiSeq 4000 platform using two HiSeq 3000/4000 SBS 50 cycles kits (Illumina). For P12, post-vaccination samples were paired-end (100 nt) sequenced on an Illumina NovaSeq 6000 platform using an Illumina NovaSeq 6000 S2 Flow Cell and an Illumina NovaSeq 6000 S2 Reagent Kit v.1.5 (100 Cycles) instead.Bioinformatics and mutation discoveryAll genomics-related data analysis steps were coordinated by proprietary bioinformatic pipeline (BioNTech) implemented in the Python programming language and described in brief below. For each of the replicate of the DNA libraries, at least 180 × 106 paired-end 50 nt reads were available covering ≥70% of targeted bases with ≥100× coverage. For the RNA libraries a minimum of 75 × 106 paired-end 50 nt reads were required.For mutation detection, DNA reads were aligned to the reference genome hg19 with bwa (v.0.7.10)32. The resulting alignment files were converted to BAM format using SAMtools (v.0.1.19)33. Somatic SNVs were called using an in-house mutation caller4 and short insertions and deletions were called using Strelka34, by comparing the aligned reads of the tumour DNA to those of the matching PBMC DNA.Genomic coordinates of identified somatic variants were compared with the UCSC Known Genes transcript coordinates to associate the variants with genes, transcripts, and potential amino acid sequence changes. Synonymous and non-sense mutations were filtered. For verified and non-synonymous cancer mutations that were selected as potential neoantigen vaccine targets, a mutated peptide sequence (MPS) was determined based on the mutated transcript sequence for contribution to design of a MPS concatemerized vaccine. In the case of SNVs, these were the 27mer peptide regions with the changed amino acid in the centre. For insertions and deletions resulting in a frameshift, the MPS featured the sequence from the changed amino acid to the next stop codon (maximum 50 amino acids). Germline variants in the region of the mutated peptides were identified using SAMtools based on the matching PBMC DNA. Protein changing germline variants were first phased based on the RNA-seq reads and, if in-phase with the somatic mutations, included in the patient-specific neoantigen vaccine target.For RNA-seq, RNA reads were aligned to the hg19 transcriptome using sailfish (v.0.7.6)33 to estimate the transcript expressions. Non-expressed transcripts were filtered.Further, RNA reads were aligned to the hg19 reference genome using STAR (v.2.4.2a)35 for phasing somatic with germline variants, as well as for determining the relative expression of a mutated transcript in comparison to the transcript not carrying the somatic mutation.NGSCheckMate (v.1.0)36 was used to confirm the identical patient as origin for all DNA and RNA libraries analysed under the same patient ID.Neoepitope prioritization and selectionHLA binding affinity was predicted via the IEDB T cell prediction tools (v.2.13)37. For HLA class I the affinity of all variant-containing 8–11mers of the MPS for HLA-A/B/C was predicted using the IEDB-recommended mode. For HLA class II, the affinity of all variant-containing 15mers of the MPS for HLA-DRB was predicted using the consensus3 method. Out of all predictions for a single variant, the best consensus score was associated with the respective MPS.Up to 46 MPS from all identified MPS were prioritized using the same in-house bioinformatics pipeline using the sorting and filters described below. First, only somatic mutations with a variant allele frequency in RNA > 0 were considered. From these: (1) up to 5 MPS from insertions or deletions were selected based on their HLA class I binding affinity; (2) up to 20 MPS from SNVs based on their HLA class II binding affinity and transcript expression ≥10 RPKM; (3) up to 20 MPS from SNVs based on their HLA class I binding affinity and transcript expression ≥1 RPKM; (4) further MPS from SNVs based on their transcript expression to reach 46 MPS in total; in case less than 46 MPS could be selected, somatic mutations with a variant allele frequency in RNA of zero were added based on (6) HLA class I score; and (6) transcript expression.An algorithm implemented in R was used to select up to 20 neoantigen vaccine targets from the list of prioritized MPS based on HLA I and HLA II binding predictions, transcript expression, variant allele frequency, and other criteria. The selection was manually reviewed per patient by a review board.Good manufacturing practice manufacturing of RNA–LPXManufacturing runs were conducted for all patients during 2019. No pre-specified target was set for turnaround time, as the individualized on-demand manufacturing process for neoantigen RNA–LPX was initiated as part of this clinical trial and continuously optimized during patient enrolment.In short, two synthetic DNA fragments each coding up to ten neoantigen vaccine targets (SNVs and short insertions and deletions) connected by non-immunogenic glycine/serine linkers (30 bp long) were cloned into a starting vector8, containing the SEC10 (SEC, MRVMAPRTLILLLSGALALTETWAGS) and the MITD domain sequences10 (MITD, IVGIVAGLAVLAVVVIGAVVATVMCRRKSSGGKGGSYSQAASSDSAQGSDVSLTA) for fusion to mutated sequence concatemers for optimized routing to HLA class I and II pathways. The starting vectors also contained the FI-elements9 (FI, LVLHARNASCPFPVLGTPSLPRPRVPGMLPPPPAPLTTSASSRHL) and backbone sequence elements for improved RNA stability and translational efficiency. The DNA was linearized via PCR, spectrophotometrically quantified, and identified with Sanger sequencing, and subjected to in vitro transcription with T7 RNA polymerase as previously described in the presence of ATP, CTP, UTP, GTP and β-S-ARCA(D1) cap analogue in a cleanroom environment. RNA was purified using magnetic particles and integrity was assessed by gel electrophoresis and microfluidic capillary electrophoresis. Further analyses included determination of concentration, pH, osmolality, potency, and endotoxin level.Liposomes with net cationic charges were used to complex the RNAs to form RNA–LPX. The cationic liposomes were manufactured using an adopted proprietary protocol38 based on the ethanol injection techniques39 from the cationic synthetic lipid (R)-N,N,N trimethyl-2-3-dioleyloxy-1-propanaminium chloride (R-DOTMA) and the phospholipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (Merck and Cie). Release analysis for the liposomes included determination of appearance, lipid concentration, RNase presence, particle size and polydispersity index, osmolality, pH, subvisible particles, pyrogen testing, and sterility. The RNA–LPX drug products were prepared in a good manufacturing practice manufacturing facility by first incubation of the individual RNAs with 14.6% (w:v) aqueous NaCl (Hospira) to arrive at the intended NaCl concentration in the RNA solution prior to mixing with the liposomes. Subsequent automated mixing of the pre-conditioned RNA with the liposomes was performed using a proprietary process, which allowed for accurate control of the mixing ratio between the two moieties. RNA–LPX were further diluted to the final drug product concentration by the addition of cryoprotectant containing customized buffers. The bulk drug product was filled into type 1 glass vials (Ompi). After inspection for visible particles, the drug product was frozen and delivered to the hospital as a concentrated dispersion for dilution. Release analysis for the RNA–LPX included determination of appearance, particle size and polydispersity, subvisible particles, RNA integrity, RNA content, pH, osmolality, endotoxin testing and sterility. For administration to the patient, the thawed drug product was diluted with 0.9% aqueous NaCl.Blood sampling for immunogenicity assaysFor assessment of vaccine-induced immune responses, blood was sampled at baseline, before the fourth, sixth and eighth vaccine dose and 7–14 days after the eighth dose. After end of treatment, blood samples were taken 49–63 days after last dose. For patients actively participating in long-term follow-up, further samples were collected every 3 months after end of the main study phase in the first year and every 6 months in the second and third year. Patients not actively participating in long-term follow-up could donate blood for research purposes via a research project during routine visits at their clinic. PBMCs were isolated by Ficoll-Hypaque (Amersham Biosciences) density gradient centrifugation from peripheral blood or leukapheresis samples.Overlapping peptidesSynthetic 15mer peptides with 11 amino acid overlaps covering the neoantigen sequences, referred to as OLP pools (>70% purity), or 8–11mer peptides (>90% purity) were used in immunogenicity assessments. All synthetic peptides were purchased from JPT Peptide Technologies GmbH and dissolved in 10% DMSO to a final concentration of 3 mM.IVS of PBMCsCD4+ and CD8+ T cells were isolated from cryopreserved PBMCs using microbeads (Miltenyi Biotec). Individual IVS cultures were set up using 15mer OLP pools encoding patient-specific neoantigen targets with up to five targets per expansion. For this, purified CD4+ T cells were expanded in the presence of fast dendritic cells, effector to target ratio (E:T) of 10:1, and the respective peptides. For the expansion of CD8+ T cells, purified CD8+ T cells were co-cultured with CD4-depleted PBMCs (E:T, 1:10 or 1:20) in the presence of IL-4 and GM-CSF (each 1.000 U ml−1) and the respective peptides. One day after starting the IVS, fresh culture medium containing 10 U ml−1 IL-2 (Proleukin S, Novartis), 5 ng ml−1 IL-15 (Peprotech) were added. CD8 IVS cultures additionally received IL-4 and GM-CSF (each 1,000 U ml−1). Seven days after setting up the IVS cultures, IL-2 was replenished (10 U ml−1). After 11 days of stimulation, cells were analysed via flow cytometry and used in ELISpot assay. Due to limited sample availability, IVS of PBMCs prior to ELISpot could only be performed for eight patients.IFNγ ELISpotMultiscreen filter plates (Merck Millipore), precoated with IFNγ-specific antibodies (Mabtech), were washed with phosphate-buffered saline (PBS) and blocked with X-VIVO 15 (Lonza) containing 2% human serum albumin (CSL-Behring) for 1–5 h. Next, 0.5 × 105 to 3.3 × 105 effector cells per well were stimulated for 16–20 h either with peptides (ex vivo setting) or with autologous dendritic cells loaded with peptides (IVS samples). For analysis of ex vivo T cell responses, cryopreserved PBMCs were subjected to ELISpot after a resting period of 2–5 h at 37 °C. Alternatively, CD4- or CD8-depleted PBMCs were used as CD8 or CD4 effectors. All tests were performed in duplicate or triplicate and included positive controls (anti-CD3 (Mabtech; 1:1,000)), Staphylococcus enterotoxin B (Sigma-Aldrich) or PHA-L (Thermo Fisher). Bound IFNγ was visualized either using a secondary antibody directly conjugated with alkaline phosphatase (ELISpotPro kit, Mabtech; for ex vivo) or biotin-conjugated anti-IFNγ (Mabtech, 1:1,000) followed by incubation with ExtrAvidin–Alkaline Phosphatase (Sigma-Aldrich) for IVS samples. Next, plates were incubated with BCIP/NBT (5-bromo-4-chloro-3′-indolyl phosphate and nitro blue tetrazolium) substrate (ELISpotPro kit, Mabtech (ex vivo) or Sigma-Aldrich (IVS samples)). Plates were scanned using either an AID Classic Robot ELISpot Reader or CTL ImmunoSpot Series S6CORE analyser (ImmunoCapture Image Acquisition Software v.6.6) and analysed by AID ELISpot 7.0 software (AID Autoimmun Diagnostika) or ImmunoSpot Professional Software v.5.4. Spot counts were displayed as mean values of each duplicate or triplicate. In the ex vivo setting, peptide-stimulated spot counts for each sample were compared to effectors from the same sample incubated with medium only, as negative control using an in-house ELISpot data analysis tool (EDA), based on two statistical tests (distribution-free resampling) according to previous publications40,41. In case of a P value of <0.05 and minimum 7 counts, response to that peptide (neoantigen) was defined as a positive response. In the next step, ELISpot data analysis tool response calls for the pre- and post-vaccination samples were compared. Neoantigens with a positive response call only in the post-vaccination sample were classified as inducing a de novo response. Two neoantigens induced a significant increase in spot counts in the pre-vaccination sample. These spot counts were >11 fold higher in the post-vaccination sample (Extended Data Fig. 3, amplified responses highlighted in green boxes). These responses were classified as ‘amplified’. In the post-IVS setting, peptide-stimulated spot counts were compared to control peptide-loaded target cells using an in-house statistical analysis tool for pre- and post-vaccination samples. T cell vaccine responses were defined with an at least twofold increase in spot count after vaccination.Peptide–MHC multimer stainingMutation-specific CD8+ T cells were identified using fluorophore-coupled peptide/MHC (pMHC) multimers (ImmunAware) carrying 9-to-11-amino-acid peptides from immunogenic neoantigens. Potential minimal class I epitopes were identified by the NetMHCpan predictor (DTU Health Tech). Alternatively, peptide–MHC complexes were generated using easYmer technology (easYmer kit, ImmuneAware Aps), and complex formation was validated in a bead-based flow cytometry assay according to the manufacturer’s instructions. For tetramerization, streptavidin (SA)–fluorochrome conjugates were added: SA–BV421, SA–BV711, SA–PE, SA–PE-Cy7 or SA–APC (all BD Biosciences). Cells were stained for multimers first and then for cell surface markers, as follows (antibody clones and dilution in parentheses): CD28 (CD28.8, 1:25), CD197 (150503, 1:50), CD45RA (HI100, 1:100), CD3 (SK7, 1:100), CD16 (3G8, 1:100), CD14 (MφP9, 1:50), CD27 (L128, 1:50), CD279 (EH12, 1:25), CD127 (HIL-7R-M21, 1:50) and CD8 (RPA-T8, 1:50), all purchased from BD Biosciences; CD19 (HIB19) and CD4 (OKT4), from Biolegend. TCF-1 (C63D9, Cell Signaling) was stained intracellularly after permeabilization. We also carried out live-dead staining using 4′,6-diamidino-2-phenylindole (DAPI; BD) or fixable viability dyes eFluor 780 or eFluor 506 (eBioscience). Singlet, live, multimer-positive events were identified within CD3+ (or CD8+), CD4−CD14−CD16−CD19− or CD3+ (or CD8+) CD4− events (Extended Data Fig. 7a). For detection of antigen-specific T cells after IVS, single, live, CD3+, CD8+multimer+ lymphocytes were gated.Intracellular cytokine stainingPBMCs were incubated with peptides for around 16 h at 37 °C in the presence of brefeldin A and monensin. Cells were stained for viability (using fixable viability dye eFluor 780, eBioscience, 1:1667) and for surface markers CD8 (RPA-T8, 1:33), CD16 (3G8, 1:100), CD14 (MφP9, 1:50) (all from BD Biosciences), CD19 (HIB19, 1:50), or CD4 (OKT4, 1:25) (from Biolegend). After permeabilization, intracellular cytokine staining was performed using antibodies against IFNγ (B27, BD Biosciences, 1:100) and TNF (Mab11, BD, 1:167). IFNγ+ and TNF+ events were identified within the CD8+ and CD4+ cells pre-gated on single, live, and CD14−CD16−CD19− (not used in all experiments) populations (Extended Data Fig. 7b).Acquisition was performed on a LSR Fortessa SORP or FACSCanto II cell analyser (BD Biosciences) and analysed via FlowJo software (Tree Star).Tumour bulk TCR profilingDNA was extracted from FFPE tumour tissue as described in ‘Next generation sequencing’. Sequencing and bioinformatic analysis to retrieve tumour TCR clonotypes was performed by iRepertoire using their proprietary pipelines.Blood bulk TCR sequencing and bioinformatic analysisTotal RNA was extracted from 3–5 × 105 CD8+ or CD4+ T cells sorted by magnetic-activated cell sorting (MACS) using RNeasy Mini Spin Columns Qiagen kit. Total RNA was eluted in 30 μl water and the total yield was used for bulk TCR sequencing. Bulk TCR profiling libraries were generated using the SMARTer Human TCR a/b Profiling kit from Clontech Laboratories (now Takara Bio), according to the manufacturer’s manual. Sequencing of the library was performed on an Illumina MiSeq sequencer using the 600-cycle MiSeq Reagent Kit v.3 with paired-end, 2× 300 base pair reads. The data were demultiplexed using the Bcl2Fastq software (Illumina). Fasta sequences were edited performing a quality trimming step using the open-source trimmomatic v.0.36 software and then analysed with the open-source MiXCR software to annotate TCR sequences. TCR clonotypes with identical complementarity determining region 3 (CDR3) at the amino acid level were merged and their count and frequency values summed together. An in-house script was used to calculate repertoire statistics. Tracking of neoantigen-specific T cell clones in profiling data over time and in tumour tissue was performed using TRB CDR3 chain amino acid sequence as a molecular identifier.Single-cell sequencing and read processingMACS-sorted CD8+ or CD4+ cells were washed once with 1 ml Dulbecco’s PBS (DPBS) with 0.04% BSA using a wide-bore tip centrifuged at 300g for 4 min, and resuspended at 5.3 × 105 cells per ml. Twenty thousand cells (37.8 µl) were then transferred into a fresh tube and libraries for TCR VDJ and gene expression analysis prepared following the manufacturer’s instruction (User Guide Chromium Next GEM Single-Cell V(J)J Reagent Kits v.1.1, 10x Genomics, CG000207 Rev. D). Sequencing of the TCR libraries were performed on an Illumina MiSeq sequencer using the MiSeq Reagent Kit, 600 cycles, v.3 with paired-end, 2× 150 base pair reads. Sequencing of the 5′ gene expression library was performed on an Illumina NovaSeq sequencer using the NextSeq 1000/2000 P2 reagents, 100 cycles, v.3 with paired-end, 26× 91 base pair reads. Raw sequencing data were processed using the Cell Ranger software (10x Genomics) to generate clonotype data and raw count matrices of gene expression. For the VDJ data cell ranger, output files were then reshaped to a compatible format with our in-house data analysis pipeline and filtered to keep only clonotypes that contain both a TRA and a TRB chain. Clonotypes with two TRB chains were discarded.RevImMo analysisBulk TCR profiling data generated from pre- and post-vaccination CD8+ (and CD4+) T cells were used to calculate clonal enrichment post-treatment for each TRB CDR3 clonotype. For de novo clones, which were not detected pre-treatment, treatment-induced enrichment was calculated by assigning an arbitrary frequency value corresponding to the lowest frequency detected in the corresponding repertoire. Clones present at both time points were termed pre-existing. Single-cell TCR profiling data from post-treatment CD8+ (or CD4+) T cells were used to retrieve the paired TRA/TRB information for each TRB clonotype. Only TRB clonotypes with paired information were ranked by enrichment and used for clone selection (Fig. 3a). Ten to fifteen each of the most enriched pre-existing and de novo TCRs were selected for further validation. Synthesized TCR V(D)J genes of the selected candidates were cloned into the pST1-TRAC/TRBC1/TRBC2 vector backbones by Twist Bioscience. DNA template manufacturing PCR was performed to amplify linearized templates of the cloned TRA and TRB chains which were then used to produce in vitro transcribed RNAs for validation of their specificity. The vaccine neoantigen specificity and functionality of the selected TCR candidates was evaluated using an optimized Jurkat-NFAT-reporter assay. The genetically engineered Jurkat T cell line expresses luciferase as a reporter, driven by an NFAT-response element (NFAT-RE), which is induced by the TCR-specific signalling cascade. NFAT–TCR/CD3 effector cells were purchased from Promega as cryopreserved cells. Reauthentication of cell lines was performed by short tandem repeat (STR) profiling at ATCC and Eurofins. All used cell lines tested negative for mycoplasma contamination. Optimization of used reporter cells was conducted by CRISPR–Cas9-mediated knockout of the endogenously expressed TCR and by transposon-based stable insertion of the CD8 co-receptor (alpha and beta chain). After electroporation of TRA- and TRB-encoding IVT-RNAs and a 20 h incubation period, 2 × 104 Jurkat cells were co-cultured with K562 cells or autologous CD14+ monocyte cells at a 5:1 ratio, in a 384-well plate with 25 μl medium (RPMI1640 + 10% non-heat inactivated FBS) per well. Prior to co-culture, the K562 cells were transfected with IVT-RNA encoding the patient’s HLA alleles and loaded with neoantigen target peptide pools. The autologous CD14 cells were loaded with neoantigen target peptide pools. For all assays, patient-specific neoantigen targets were tested on all patient-specific HLA alleles (Extended Data Fig. 4). After 6 h, an equal volume (25 μl) of luciferin (Bio-Glo, Promega) was added to each well and the luciferase activity was measured using a luminescence plate reader. The measured luminescence signal in the different wells corresponded to the level of TCR-mediated activation in the Jurkat cells. For each TCR, fold change of luminescence compared with the TCR-transfected effectors-only control was calculated and a cut-off of twofold change was used to determine specific TCRs. In selected cases, negatively tested TCRs were subsequently evaluated by multimer-based staining experiments. CD8 TCR no. 18 from P1 was determined positive using this method.Single-cell gene expression analysisCount matrices were analysed using Seurat software v.5.1.042. Cells were associated with the corresponding VDJ data using cell barcodes and only cells with an associated VDJ clonotype were kept. To eliminate low quality cells and doublets, cells expressing two different TRB chains, more than 6,000 and less than 100 unique genes and with a percentage of mitochondrial unique molecular identifiers, more than 10% were excluded from the analysis. Filtered data were then normalized and scaled using the SCTransform function of the Seurat package43 with default parameters and regressing the percentage of mitochondrial counts. Data filtering and normalization was performed for each sample separately. Normalized data from different time points and patients were then integrated using Harmony 1.2.1 using RunHarmony function. After performing principal component analysis, the first 14 principal components were selected by examining the ‘elbow plot’. Clustering analysis was performed using the FindNeighbors and FindClusters functions with a resolution of 0.5. Cluster identity was assigned based on differential expressed genes identified using FindAllMarkers Seurat function. Based on TRB CDR3 sequences, neoantigen-specific cells were identified and selected for further analysis. These cells were reanalysed as described above using the first 10 principal components and a resolution of 0.3. After differential gene expression analysis, clusters with similar phenotype were merged.Single-cell TCR sequencing of multimer-positive cellsThree peptide–HLA dextramer reagents were prepared by mixing each 2.16 pmol peptide-loaded HLA monomer (Immudex) with 0.48 µl ULoad dCODE dextramer, conjugated to a specific DNA barcode (Immudex). Peptide–HLA monomers and dextramers were incubated for 30 min in the dark at 4 °C. An empty dextramer was used as a control. For multimer staining, 0.8 µl 100 µM biotin was combined with assembled peptide–HLA and control dextramers for pooled staining of PBMCs. PBMCs were resuspended and washed in MACS buffer. A total of 10 × 106 viable PBMCs was resuspended in PBS/2% human serum albumin (HSA) and stained with 16.6 µl dextramer pool after adding 5 µl Human TruStain FcX Block (Biolegend) in a total volume of 50 µl. Cells were stained for 10 min at 4 °C in the dark. Fifty microlitres antibody surface staining mix was added (BV480 mouse anti-human CD8, BB515 mouse anti-human CD4, APC-eFlour780 mouse anti-human CD16/CD14/CD19, fixable viability dye eFlour780). Cells were stained with antibody mix for 30 min at 4 °C. Cells were then washed 3 times with each 1 ml PBS/2% HSA and resuspended in a volume of 500 µl PBS/2% HSA. Multimer-positive cells were sorted by fluorescence-activated cell sorting (FACS) by first gating the dump channel (eFlour780)-negative, CD8-positive lymphocyte population. Dextramer backbones were phycoerythrin (PE)-labelled such that multimer-specific CD8 T cells were isolated as the PE-positive population. Sorted multimer-positive T cells were centrifuged and resuspended in a volume of 50 µl in PBS/0.04% BSA. Cell concentration was determined, and cells were diluted to a final concentration of 5.3 × 105 cells per ml. Single-cell RNA-seq was performed as described above. VDJ and Feature Barcoding libraries were produced as described by the manufacturer’s instructions (Chromium Next GEM Single-Cell 5′ Reagent Kits v.2 [Dual Index], 10x Genomics, CG000511 Rev. B). Data Processing of VDJ libraries was performed as described above. Feature barcoding libraries were processed using Cell Ranger software to assign multimer barcodes to sequenced cells. Antigen specificity of individual TCR clones was determined using in-house scripts after merging VDJ and feature barcoding data based on cell barcodes.Immunohistochemical stainingTo determine the expression of CD3, CD8 and MHC-I, 3- to 4-µm-thick sections of FFPE tumour tissue were analysed.Staining was performed on the Ventana Discovery Ultra platform. For antigen retrieval Ultra CC1 was used for 64 min. Slides were afterwards incubated for 60 min at 37 °C with anti-human CD3 (2GV6; Roche Diagnostics at a ready-to-use dilution), anti-human CD8 (SP57; Roche Diagnostics, 1:100) and MHC-I (EPR1394Y; Abcam, 1:500) followed by secondary antibody OmniMap anti-rabbit HRP (Roche Diagnostics, at a ready-to-use dilution) for 32 min at 37 °C. For visualization, ChomoMap DAB Kit (Roche Diagnostics) was used followed by counterstain (Hematoxylin II, Bluing reagent; Roche Diagnostics). Slides were scanned (Axio.Scan; Zeiss) and manually analysed.Differential gene expression analysis, GSEA and variant allele frequency calculationWhole-exome sequencing and RNA-seq data for P12 and P13 from post-treatment samples were analysed using the bioinformatics software pipeline. Differential expression analysis on RNA-seq data was performed with R (v.4.0.2) using tximport (v.1.18) to load read counts generated by sailfish and summarize transcript read counts by genes. Summarized counts were imported into DESeq2 (v.1.30)44 for differential expression testing according to instructions on the package vignette, only considering genes that were covered by at least ten reads across all samples. Differential expression testing results were exported with a significance cut-off of 0.01 (after false discovery correction using the Benjamini and Hochberg method45) and shrunken log fold change values were calculated. GSEA was performed using fgsea 1.20.046 R package on MSigDB v.7.5.147 KEGG canonical pathways. Pre- versus post-treatment tumour differential expressed genes were ranked based on the Wald statistic value and used as input of the fgsa function performing 1,000 permutations. Variant allele frequencies for somatic SNVs called in the pre-treatment tumour were determined from the aligned tumour BAM files using pysam (v.0.15.4)48,49,50.Kaplan–Meier time-to-event analysis for disease-free survival probabilityDisease-free survival was a non-protocol-specified analysis performed for this report. Disease-free survival was defined as the time from first vaccine dose to cancer recurrence or death from any cause. Imaging after initial (neo)adjuvant treatment was only performed upon suspected progression, which may overestimate disease-free survival. Date of last visit/contact served as the date of censoring. The analysis was performed using SAS v.9.4.Materials availabilityMaterials are available from the authors under a material transfer agreement with BioNTech. This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. This license does not apply to figures/photos/artwork or other content included in the article that is credited to a third party; obtain authorization from the rights holder before using such material.Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.